
Enfermedades
Neurodegenerativas: el Caso de la Proteína
MeCP2 y el Síndrome de Rett
Neurodegenerative diseases: The case of the MeCP2
protein and the Rett syndrome
1Rebeca Toledo Cárdenas, 1Luis
Isauro García Hernández, 2Marta
Miquel Salgado-Araujo,
1Ma. Elena Hernández Aguilar, 1Jorge
Manzo Denes
1Proyecto Mirrus, Instituto de Neuroetología,
Universidad Veracruzana
2Área de Psicobiología, Universidad Jaume
I, Castellón, España
RESUMEN
Las
enfermedades neurodegenerativas son aquellas que se
distinguen por ser el resultado de una muerte progresiva
de neuronas en el sistema nervioso, fundamentalmente
en el cerebro. En esta revisión, se presenta el caso
del gen mecp2, que codifica para la proteína
del mismo nombre: MeCP2. Las mutaciones de este gen
y, por tanto, las malformaciones de la proteína llevan
a un tipo principal de enfermedad neurodegenerativa:
el Síndrome de Rett. Este síndrome es un padecimiento,
al parecer, exclusivo del humano, que se desarrolla
aparentemente sólo en niñas, cuando éstas tienen alrededor
de dos años de vida. La enfermedad se caracteriza por
una regresión en el desarrollo de la niña, seguida por
una pérdida de funciones motoras y cognitivas. Esto
es acompañado por alteraciones gastrointestinales severas
y por la aparición de crisis epilépticas. Este padecimiento
es prolongado, por lo que las niñas fallecen generalmente
después de la pubertad, lo que indica que el paso por
su etapa de infancia es en un estado devastador. Este
padecimiento tiene muchas características similares
al autismo, por lo que varios investigadores lo consideran
un caso especial del mismo. La causa del Síndrome de
Rett es una mutación en el gen mecp2, que evita la síntesis adecuada
de la proteína MeCP2. Esta proteína es represora de
otros genes, y su ausencia hace que esos genes se expresen
sin control, lo que lleva a la aparición de la patología.
Aún se desconocen cuáles genes son los regulados por
la proteína MeCP2, por lo que un acercamiento terapéutico
para aliviar los malestares en estas niñas aún está
lejos de encontrarse. Esta revisión tiene el propósito
de analizar someramente este caso, que ha sido el punto
de partida del proyecto iniciado recientemente en nuestra
dependencia de
Palabras
clave: cromosoma X, mutación, gen MECP2, Metilación, Islotes CpG, Autismo
ABSTRACT
Neurodegenerative diseases
are the result of a progressive death of neurons in
the nervous system, mainly of those located in the brain.
This review deals with the specific case of the mecp2 gene that encodes the protein with
the same name: MeCP2. Mutations at this gene and, therefore,
disfunction of the protein lead to a specific kind of
neurodegenerative disease: the Rett Syndrome. This syndrome
seems to be specific of humans and develop mainly in
girls around two years of age. The disease is characterized
by a regression in the development of the girl, followed
by the lost of motor and cognitive functions. Furthermore,
there are other accompanying misfunctions as severe
gastrointestinal modifications and epileptic seizures.
A major concern is the long lasting effects of this
neurodegeneration, since girls die once they arrive
to puberty; thus, their infant period is lived in a
devastating situation. Rett syndrome has a number of
responses similar to those observed in autism, hence
some authors has classified it as a special kind of
itself. The cause of the syndrome is the mutation at
the mecp2 gene,
which decreases the appropriate synthesis of the MeCP2
protein. This protein is a gene repressor, i.e., when
absent several genes became activated without control,
triggering the appearance of the syndrome. Still we
do not know which genes are regulated by the MeCP2 protein,
hence a therapeutic approach to alleviate the sickness
of the girls is far from being discovered. This review
has the purpose to analyze this case that served as
the starting point of the project at the Universidad
Veracruzana: the Mirrus Project for the Study of Development
Disorder of Neurodegenerative Diseases.
Las enfermedades neurodegenerativas son aquellas que
se distinguen por ser el resultado de una muerte progresiva
de neuronas en el sistema nervioso, fundamentalmente
en el cerebro. La muerte de estas células, aunada a
que están inhabilitadas para duplicarse, colocan al
individuo en un estado constante de pérdida de funciones,
que va desde incapacidad de movimiento corporal hasta
la alteración de facultades mentales. Esta degeneración
del sistema nervioso es el resultado de diferentes tipos
de etiologías; puede ser inducida por agentes externos
como virus y otros poco convencionales como los priones,
o puede ser intrínseca del sujeto como resultado de
mutaciones genéticas. Las mutaciones son las más comunes
y, dependiendo de los genes involucrados, producen muerte
neuronal en distintas regiones del sistema nervioso,
lo que lleva a tener diferentes tipos de enfermedades
neurodegenerativas. En esta revisión, nos enfocaremos
al caso del gen mecp2,
que codifica para la proteína del mismo nombre: MeCP2
(la letra E se modifica para diferenciar el gen de la
proteína; más abajo se explica el significado de estas
siglas). Las mutaciones de este gen y, por tanto, malformaciones
de la proteína llevan a un tipo principal de enfermedad
neurodegenerativa: el Síndrome de Rett.
Las células del cuerpo tienen una organización característica
que de manera gruesa permite distinguir un citoplasma
y un núcleo. Dentro del núcleo, se distinguen los cromosomas
que, en el caso del humano, son 46. Estos cromosomas
se organizan en pares, uno proveniente del óvulo y otro
del espermatozoide, que se unen durante la fecundación,
por lo que tenemos 23 pares de cromosomas. Todos los
pares son idénticos excepto uno, el par de cromosomas
sexuales que no son iguales. Uno de los cromosomas es
denominado X y el otro Y. La organización de los gametos
en este sentido es que los óvulos siempre tienen el
cromosoma X. Los espermatozoides, por su lado, existen
en dos grupos, espermatozoides con el cromosoma X y
espermatozoides con el cromosoma Y. La unión de un óvulo
con un espermatozoide X origina un individuo XX que
se desarrolla en sexo femenino. La unión del óvulo con
un espermatozoide Y da origen a un individuo XY que
se desarrolla en sexo masculino. Así, un común denominador
entre sujetos femeninos y masculinos es que ambos comparten
al cromosoma X.
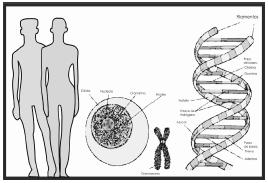
Figura 1. La información genética de un
individuo está determinada por la combinación de los
genes de cada uno de los padres, y esta información
esta almacenada en el dna
que se empaqueta
en los cromosomas, y éstos a su vez, se encuentran en
el núcleo de las células.
El gen mecp2
está compuesto por cuatro exones (secuencias particulares
dentro del gen), de los cuales el 2,3 y 4 contienen
la región codificante para la proteína, con una secuencia
de aproximadamente 1,500 pares de bases2.
El exon 1 forma parte de la región promotora del gen.
La expresión del gen mecp2 es ubicua,
se expresa en todos los tejidos, pero se ha mostrado
que da lugar a un único producto proteico, la proteína
MeCP2.
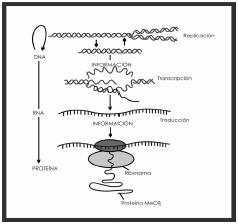
Figura 2. Flujo de
información de dna a rna
y proteínas. La
información pasa de una molécula a otra mediante el
proceso de replicación. El dna se copia en RNAm
en el núcleo, en un proceso llamado transcripción. Posteriormente
la información contenida en el ARNm es empleada para
construir proteínas en el proceso de traducción.
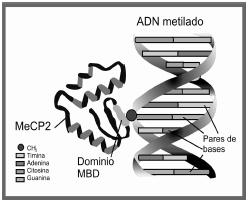
La proteína MeCP2 constituye una cadena sencilla
de 486 aminoácidos. Forma parte de una familia de proteínas
(mbd1, mbd2,
mbd3,
mbd4 y MeCP1),3 que poseen un dominio funcional característico formado
por 80 aminoácidos, conocido como methyl
CpG binding domain o dominio para la unión con CpG
metilados. Muchos tipos de genes dentro de diferentes
cromosomas tienen regiones particulares denominadas
Islotes CpG, que son secuencias con un alto porcentaje
de peldaños o pares de base C-G (la p indica la unión
fosfodiester entre C y G). Los islotes pueden sufrir
un proceso conocido como metilación, producido por una
proteína metiltransferasa del dna.4
La proteína MeCP2 se une a los islotes CpG metilados
y, como consecuencia, inhibe la expresión de esos genes,
por lo que su nombre es proteína que se une a regiones
metiladas CpG (methyl
CpG binding protein, denominada con sus siglas MeCP2,
y el 2 para diferenciarla de
El papel de
Este
padecimiento fue descrito por primera vez en 1966 por
el pediatra austríaco Andreas Rett, quien observó un
marcado proceso de retroceso, de tipo motor y cognitivo,
en niñas que parecían haber tenido un desarrollo aparentemente
normal hasta aproximadamente los 18 meses de edad.7 El Síndrome de Rett es un
desorden neurodegenerativo que parece afectar principalmente
a niñas con una incidencia aproximada de 1 caso en
En la mayoría de los casos, la aparición del Síndrome de Rett es por la
mutación esporádica del gen mecp2,
aunque existen algunos casos en que el gen ya estaba
mutado en alguno de los padres y así es transferido
a la descendencia. Sin embargo, las alteraciones exactas
que una niña puede tener aún son difíciles de determinar.
Esto porque las mutaciones al gen mecp2
pueden ser de diversos tipos, desde mutaciones en un
exon diferente, del 1 al 4, que pueden ser supresiones
del número de pares de bases o adiciones de pares de
bases. Cualquiera de estos mecanismos lleva a un producto
parecido pero distinto a la proteína MeCP2 normal, pero
que modifica la alteración de genes que a la fecha son
difíciles de seguir.
Una pregunta que desde el
principio surgió es por qué sólo las niñas. Las primeras
hipótesis sugirieron que la organización xx
del par de cromosomas sexuales les confería cierta protección
a las niñas (al tener dos genes mecp2,
uno en cada cromosoma), con respecto a los niños que,
al ser xy, y por tanto tener sólo un gen, son más vulnerables. Esta
hipótesis no pudo ser probada hasta la aparición de
modelos animales, como algunos ratones. Actualmente existen tres modelos predominantes
de ratones modificados genéticamente que presentan una
mutación en el gen mecp2
y desarrollan patrones similares al Síndrome de Rett
en humanos. Se ha observado en estos animales,12
que la alteración del gen en machos es letal a nivel
embrionario, por lo que se sugiere que la presencia
de niños Rett es extremadamente rara dada su baja sobrevivencia
en etapa embrionaria; esto es, no llegan a nacer. Sin
embargo, se han encontrado ya algunos niños Rett, en
los que se desconoce la causa de su relativa sobrevivencia,
pero que presentan síntomas de deterioro mental mucho
más severos que las niñas. Esto ha llevado a proponer
que el grado de mutación del gen mecp2
parece estar correlacionado con la severidad del síndrome,
aspecto que aún se encuentra bajo investigación.13
La mutación del gen mecp2, con la consecuente alteración en
la síntesis de la proteína MeCP2, tiene como resultado
inicial una regresión en el crecimiento del cuerpo de
las neuronas, una reducción en el tamaño de las dendritas
y una reducción de la capacidad de estas neuronas de
establecer contactos sinápticos, que eventualmente llevan
a la muerte de las neuronas. Como consecuencia, el tamaño
de las regiones cerebrales en donde se observan estos
cambios se ve reducido. Así, se observan reducciones
significativas en el hipocampo, la corteza cerebral
y el cerebelo. Estas tres estructuras ocupan lugares
centrales en el establecimiento de funciones cognitivas
y motoras, por lo que sus alteraciones son las que llevan
a todas las características observadas en sujetos Rett14.
El Síndrome de Rett expresa
muchas características similares al autismo, por lo
que ha sido agrupado dentro del patrón de enfermedades
autistas. A la fecha, se ha observado que en un alto
porcentaje de personas autistas, la alteración en la
expresión de la proteína MeCP2 también se encuentra
presente. Considerando todas las alteraciones funcionales
y conductuales que se presentan tanto en el autismo
como en el Síndrome de Rett, una de las hipótesis que
se tiene actualmente es que la proteína MeCP2 tiene
la función de silenciar genes directamente relacionados
con la conducta social de los individuos. Sin embargo,
esto está aún lejos de ser investigado.
- Wan M, et al. Rett síndrome and beyond: Recurrent spontaneous and familial mecp2 mutations at CpG hotspots.
Am. J.
Hum. Genet 1999; 65: 1520-9.
- Nagai K, Miyake K, Kubota T. A
transcriptional repressor MeCP2 causing Rett syndrome
is expressed in embryonic non-neuronal cells and
controls their growth. Develop Brain Res 2005; 157: 103-6.
- Nakao M, et al. Regulation of transcription and crhomatin by methyl CpG binding protein
mbd1.
Brain Dev. 2001; 23: 174-6.
-
- Boyes J, Bird A. dna methylation inhibits transcription
indirectly via a methyl-CpG binding protein. Cell 1991; 64: 112334.
- Christodoulou J, Weaving LS. MeCP2
and Beyond: phenotype-genotype correlations in Rett
Syndrome. J Child Neurol. (2003). 18: 669-74.
- Bienvenu, T, et al. The Incidence of Rett Syndrome.
Pediatric
Neurology 2006; 372-4.
- Takagi N. The role of X-chromosome
inactivation in the manifestation of Rett syndrome. Brain Dev 2001; 23: 182-5.
- Leonard H, et al. Genotype and early development in Rett syndrome:
The value of international data. Brain Dev 2005; 27: 59-68.
- Moretti P, Zoghbi H. MeCP2 dysfunction
in Rett síndrome and related disorders. Curr Opin Genet Dev 2006; (16) 3: 276-81.
- Sung Jae lee S, Wan M, Francke
U. Spectrum of mecp2 mutations in Rett syndrome. Brain Dev. 2001; 23: 138-43.
- Moog U, et al. mecp2 mutations are an infrecuent cause of mental retardation associated
with neurological problems in male patients. Brain
Dev. 2006; 28: 305-10.
- Shahbazian M, Zoghbi H. Rett syndrome and MeCP2: linking
epigenetics and neuronal function. Am J Hum Genet 2002; 71:1259-72.